Solving Biology’s 50-Year-Old Protein Puzzle
Jumper, J., Evans, R., Pritzel, A., ... & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583-589.
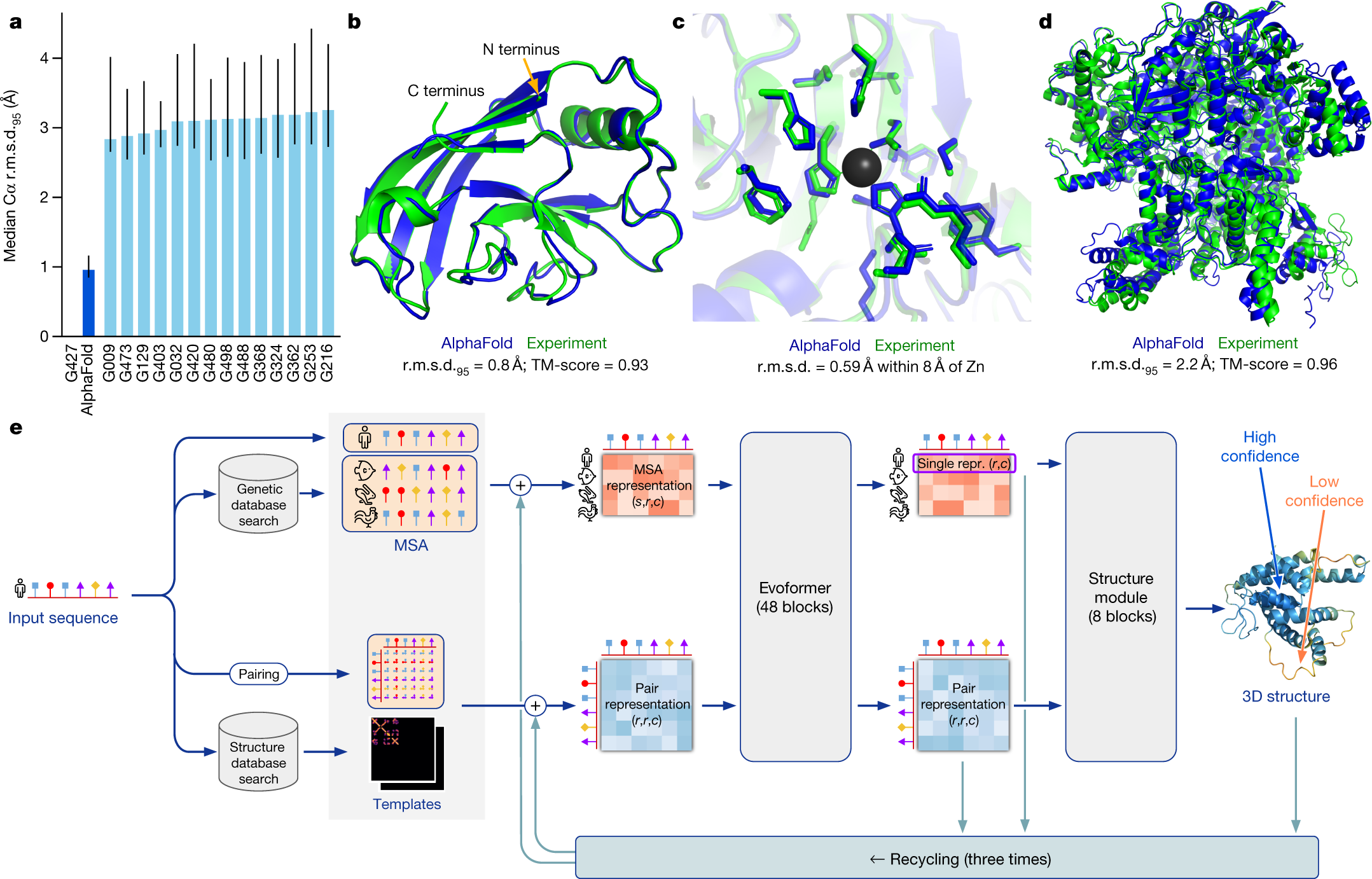
In 2021, researchers at DeepMind resolved the protein folding problem, a fifty-year grand challenge in biology, by treating the interaction of amino acids as a spatial graph problem solvable through end-to-end differentiable refinement. Prior to this research, predicting the 3D structure of a protein from its linear amino acid sequence was seen as a trade-off between the speed of statistical templates and the agonizingly slow precision of molecular dynamics simulations. AlphaFold 2 demonstrated that by utilizing an integrated attentive engine to process evolutionary and spatial constraints simultaneously, a model can achieve atomic-level accuracy across the proteome, transforming biological research from an observation-based field into a predictive science.
Geometric Refinement and the Evoformer Module
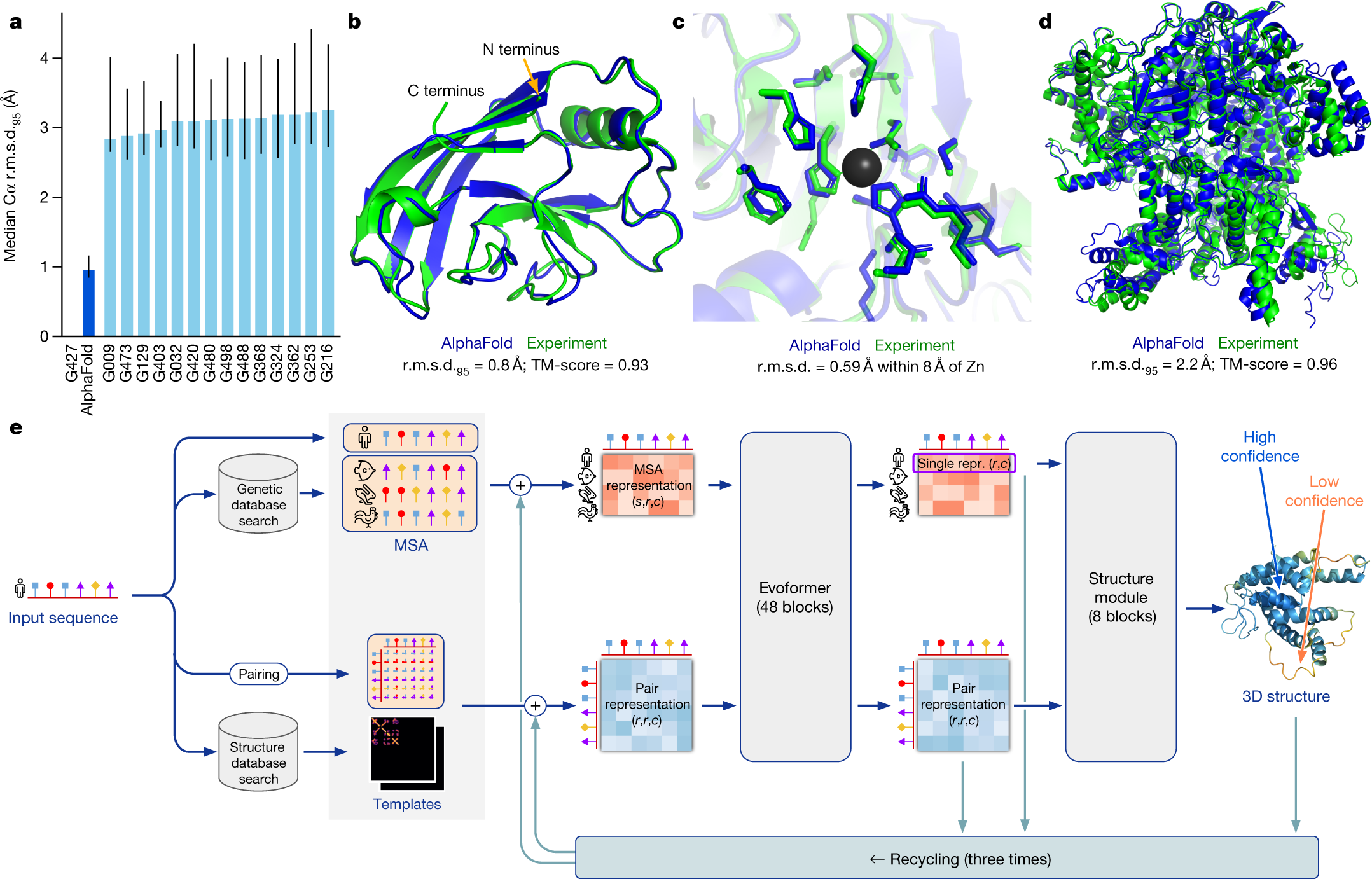
The core technical innovation of AlphaFold 2 is the Evoformer module, a specialized Transformer block that simultaneously processes two distinct types of information: the Multiple Sequence Alignment (MSA) and a "pair representation" of residue-to-residue relationships. Instead of treating evolutionary history and spatial geometry as separate features, the Evoformer uses axial attention to let them inform each other in a continuous loop. This allows the model to use the conservation patterns identified in evolution to narrow down the possible spatial configurations, while the emerging geometry of the pair representation helps filter out noise in the evolutionary data. This methodological choice established that biological information is inherently multidimensional, and that a model's performance scales when it can treat the "why" of evolution and the "how" of physics as parts of a single, unified reasoning process.
Invariant Point Attention and Physical Consistency
To maintain physical consistency without being tied to a specific coordinate system, the model utilizes Invariant Point Attention (IPA). Each amino acid residue is treated as an independent rigid body in a 3D gas, defined by a local frame of reference. The IPA mechanism allows these residues to attend to one another in a way that is mathematically invariant to the global rotation or translation of the entire protein. This approach allows the model to reason about the relative orientation and position of every part of the structure simultaneously. This finding proved that for a machine to understand physical reality, it must be equipped with architectural inductive biases that respect the fundamental symmetries of the three-dimensional world.
Atomic Accuracy and the GDT Benchmark
The technical significance of AlphaFold 2 was validated during the CASP14 competition, where it achieved a median Global Distance Test (GDT) score of 92.4 across all targets. A score above 90 is considered competitively equivalent to experimental methods such as X-ray crystallography or cryo-electron microscopy. The researchers demonstrated that the system can predict the coordinates of side-chain atoms with an error margin often less than the width of a single atom. This achievement revealed that the physical rules governing protein folding are sufficiently captured by the model's geometric engine to bypass the need for traditional physics-based energy minimization. It establishing the principle that complex biological machines can be reconstructed through the systematic management of structural information.
Impact on Structural Biology and Drug Discovery
The practical significance of AlphaFold 2 is evidenced by its role in populating the AlphaFold Protein Structure Database, which now contains predicted structures for nearly all known proteins. By providing high-fidelity models for researchers worldwide, the technology has accelerated the study of rare diseases, the development of vaccines, and the design of novel enzymes for plastic degradation. This application digitalized the act of structural discovery, replacing months of laboratory work with seconds of computation. The success of this model proved that the bottleneck in structural biology was the lack of an architecture capable of representing the complex, non-linear dependencies of a folding chain.
Structure as a Learnable Geometry
The achievement of AlphaFold 2 demonstrated that the complexity of biological systems is mathematically accessible through the lens of geometric deep learning. The decision to model folding as a differentiable refinement revealed that the primary constraint on biological intelligence was the reliance on local, sequence-only information. This principle remains the central theme in the search for "AlphaFold-like" breakthroughs in other domains, including RNA folding and small-molecule docking. It leaves open the question of whether the same geometric primitives can be used to simulate the dynamic, real-time behavior of proteins within the crowded environment of a living cell.
Join the EulerFold community
Track progress and collaborate on courses with students worldwide.
Dive Deeper
AlphaFold 2 Nature Paper (Official)
Nature • docs
Explore ResourceAlphaFold Protein Structure Database
DeepMind/EBI • docs
Explore ResourceDeepMind AlphaFold Blog
DeepMind • article
Explore ResourceCASP14 Results and AlphaFold (Video)
YouTube • video
Explore Resource
Discussion
0Join the discussion
Sign in to share your thoughts and technical insights.
Loading insights...
Recommended Readings
The author of this article utilized generative AI (Google Gemini 3.1 Pro) to assist in part of the drafting and editing process.